There are several non-structural parameters that need to be taken to convergence when using the R-A representation in photoelectron diffraction simulations: the cluster size ncluster, the multiple-scattering order nmax, the R-A order |m|max, and the maximum angular momentum lmax. In order to carry out such converged calculations most efficiently, we also include or exclude paths based on their relative importance through a control parameter pathcut, as explained in more detail below. All of these parameters except the R-A order also occur in the exact cluster methods. In this section, we address the importance of each of these parameters through a series of calculations. By default, the following quantities were chosen for all the calculations, with variations as specified in subsequent subsections.
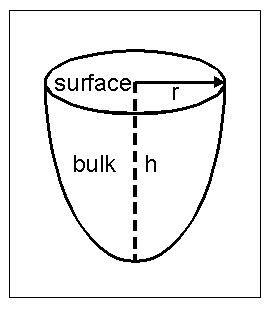
The calculations are performed for a default cluster of 86 atoms representing the ideal clean Cu(111) surface, using a lattice constant of 3.615 Å. Intensities are thus summed over emitters in various layers inward from the surface. The cluster shape is chosen to be a half ellipsoid, as shown in Fig. 6, with r = 7 Å and h = 8 Å and the emitter in each layer being positioned as close as possible to the lateral center of it. Scattering phase shifts and radial matrix elements are calculated from a muffin-tin potential due to Moruzzi et al, which uses a 1.26 Å muffin-tin radius; this leads to lmax » 7 and 13 for electron kinetic energies of 100 eV and 400 eV, respectively. The inelastic attenuation length is calculated using the TPP-2M formula of Tanuma, Powell, and Penn, which yields about 4.4 Å at an energy of 100 eV and 8.5 Å at 400 eV. No thermal vibrational damping effect is included. The inner potential was assumed to be 0 eV for these model calculations, thus neglecting any photoelectron refraction effects in crossing the surface barrier. Of course, in any actual comparisons with experiment, this parameter should be set to some reasonable non-zero value. A linearly polarized light source illuminates the surface along a [110] azimuth and at a grazing incidence angle of 10° (i.e. 80° from normal) so that the polarization lies nearly along the surface normal. Photoemission signals are taken from the Cu-3p initial state. By default, we set nmax = 8, the R-A order = 4 (15x15 matrices), lmax = 20, and the pathcut (to be defined more quantitatively below) = 0.01. Unless otherwise noted these are the values used in all calculations to systematically study parameter choices.
|
|
| Fig. 6. Cluster shape selection using a half ellipsoid. The side view is a semi-ellipse with minor axis r and major axis h. The top view is a circle with radius r. For the 86-atom cluster used to simulate Cu(111), h was 8 Å or 4 emitter layers in depth, and r was 7 Å. |
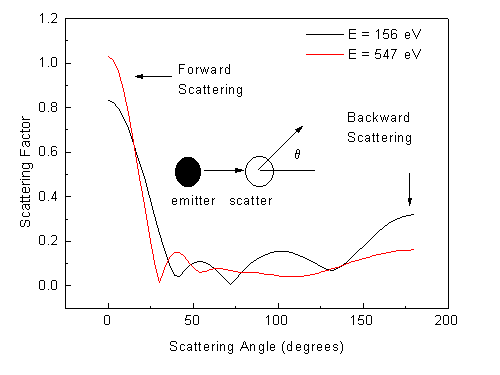
In general, photoelectrons are scattered less to large scattering angles than near forward scattering (scattering angle = 0), as illustrated for energies of 156 eV and 547 eV for a single Cu scatterer in Fig. 7. This implies that multiple forward scattering along dense rows of atoms in a crystal can be particularly strong, e.g. for deeper emitters farther from the surface of the cluster. In order to explore the influence of scattering order, the photoelectron detector is thus placed to receive electrons emitted 35.23° off the [111] normal and along a [110] nearest-neighbor forward-scattering direction in the fcc lattice. No angular broadening due to the effective detector aperture was included and kinetic energies of 156 and 547 eV (wave numbers k = 6.4 and 12.0 Å-1) were studied.
|
| Fig. 7. Cu elastic scattering factors versus scattering angle q at photoelectron kinetic energies 156 and 547 eV (wave numbers 6.4 and 12.0 Å-1). These scattering factors are calculated via the spherical-wave method for a 2-atom cluster with 2.5 Å distance. |
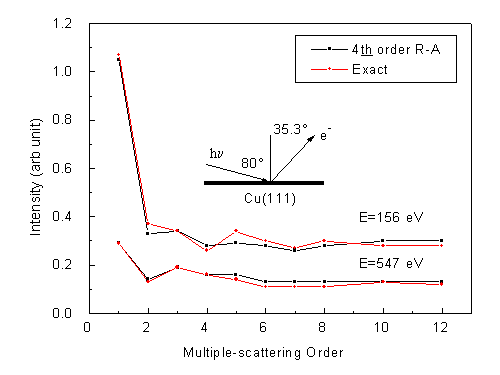
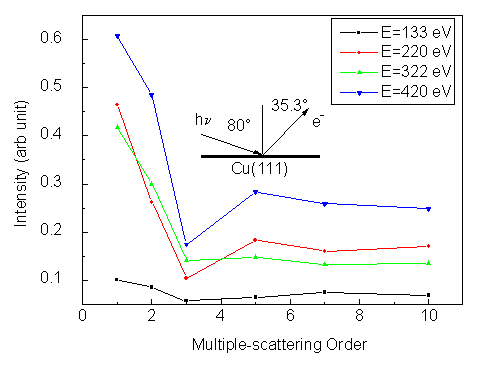
Fig. 8 shows the convergence of photoelectron diffraction intensities as a function of multiple scattering orders, for two typical energies, calculated with (|m|+2n) max = 4, i.e. in 4th order of R-A, (15x15) matrix, as compared with exact (i.e. non-R-A) calculations for the same conditions. In these calculations, the emitter is located in the third layer, so that the maximum number of forward scatterers along the direction chosen is two. A photoelectron can thus be scattered once or twice consecutively along the forward-scattering path from emitter to detector. After the 6th order of multiple scattering, the intensities have essentially converged within a few percent, and the 4th-order R-A is essentially identical to the exact results. The same sort of convergence can also be seen in Fig. 9 when the emitter is moved into the 4th layer and the forward-scattering path has three scattering atoms; here, the 7th order appears necessary to insure convergence. No exact results are shown here due to prohibitive computational times with the non-optimized program utilized. Earlier test calculations by Kaduwela et al. with the R-A methods investigated multiple scattering along long straight chains of atoms and showed similar results. The addition of thermal vibration effects should also reduce off-forward scattering amplitudes, thereby aiding the convergence of the multiple scattering series. Finally, any experimental angular averaging will tend to smear out sharper diffraction features associated with longer path length differences, further acting to enhance convergence.
|
| Fig. 8. Calculated Cu 3p photoelectron intensities, as a function of multiple scattering order, from clean Cu(111) in a fixed forward-scattering emission direction, 35.23° off-normal, and for emission from the third layer. A photoelectron can be scattered once or twice consecutively along this forward-scattering path. Photoelectron energies are 156 eV and 547 eV. Default values are used for other parameters. The 4th order R-A is compared with exact calculations. |
|
| Fig. 9. As Fig. 8, but for emission from the fourth layer. A photoelectron can be scattered up to three times consecutively along this forward-scattering path. Four different photoelectron energies are considered. No exact results were calculated. |
We thus conclude that 4th order R-A is essentially equal to exact, and that the maximum order of scattering needed to adequately simulate PD patterns is 6th or 7th, but probably lower than this with the inclusion of vibrational effects and angular averaging. This agrees with prior studies which have generally concluded that going to 4th or 5th order is sufficient.
To perform a stringent test of the R-A order, we choose a cluster of 2 Cu atoms with an arbitrary small 2.0 Å bond length (somewhat less than the actual 2.56 Å nearest-neighbor distance in Cu), because it provides the maximum sensitivity to different approximation orders. Calculations using a more realistic bond length would converge more rapidly. Closely connected to the required R-A order is the dependence of the R-A approximation on the initial state angular momentum: the 2-atom cluster also provides a good test of this question. A variety of calculations showed that low energies and single scattering are sufficient for this investigation.
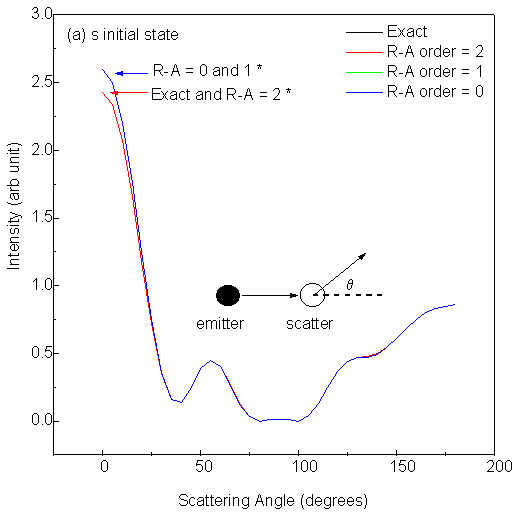
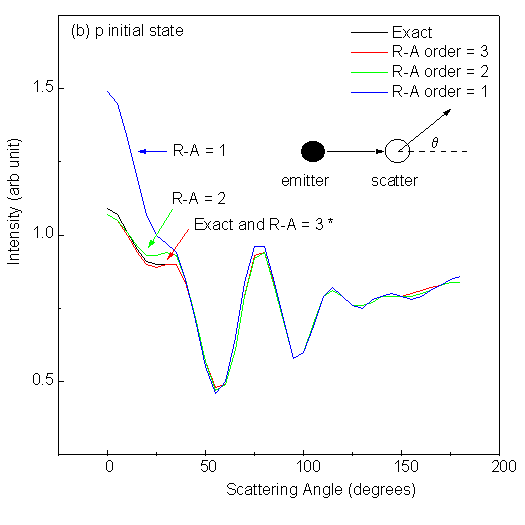
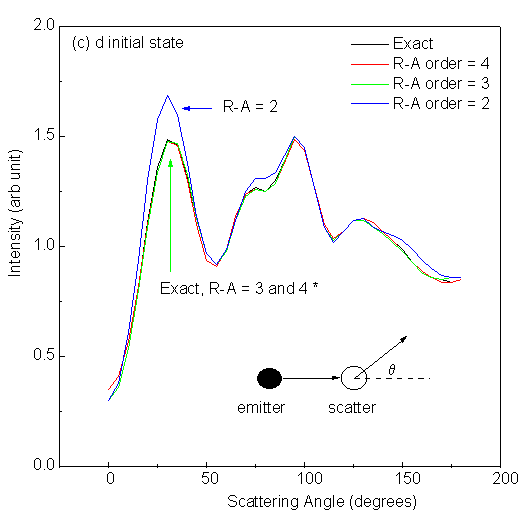
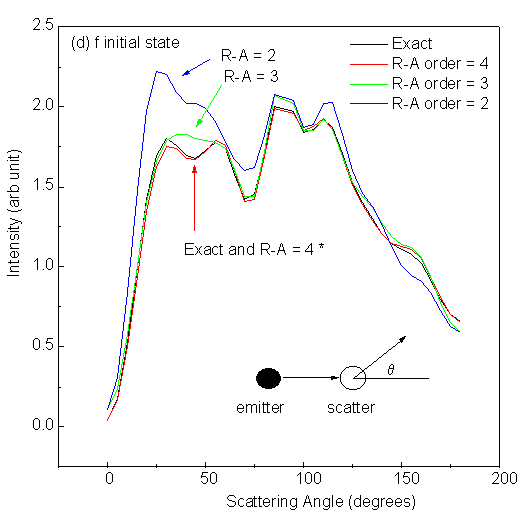
Fig. 10 shows scanned-angle photoelectron diffraction intensities for low-energy electrons (E = 61 eV) as a function of scattering angle away from the interatomic axis in the 2-atom cluster. The polarization of the light is here taken to be along the interatomic axis, in order to correspond to those atoms which are in general illuminated most strongly by the primary outgoing wave. Here q = 0° means forward scattering, while q = 180° corresponds to backward scattering. The four panels in Fig. 10 correspond to excitation from different initial states: s (li = 0), p (li = 1), d (li = 2) and f (li = 3); and in each panel we compare different R-A orders with exact results under the same conditions. It is seen that for an s initial state emitting into p photoelectron waves, the first R-A order ((3x3) matrices) is adequate and essentially identical to exact. For a p initial state emitting into s and d waves, the second R-A order (6x6) is sufficient. For a d initial state emitting into p and f waves, the third R-A order (10x10) might be needed. And for an f initial state emitting into d and g waves, the fourth R-A order (15x15) is necessary to obtain results accurate within 1%. Here, it was found that changing the initial angular momentum had a strong effect on the diffraction patterns for a two-atom cluster, but the effect of R-A order was not considered. As we shall discuss later, in a larger cluster, subsequent scatterings can be treated with equal or, more frequently lower, order in R-A, so that the R-A order needed for the first scattering is an upper limit for the entire multiple-scattering problem, and does not indicate the real limit on computing time for a given problem.
|
| Fig. 10. Single scattering intensities from a 2 Cu-atom cluster with 2.0 Å interatomic spacing and for scattering angles varying between forward (0°) and backward scattering (180°). Emission from four different initial states, s, p, d and f, is considered in panels (a), (b), (c) and (d), respectively. Default values are used for other parameters. Curves labeled with * in each panel are visually identical. |
Overall, we thus suggest a simple rule of thumb for guaranteeing adequate results: for emission from an initial state li, use the (li+1)-th R-A order for the first scattering event after emission. Other subsequent events will generally require lower orders, as dealt with in more detail in the next section.
Our MSCD R-A codes include the ability to neglect multiple-scattering paths that contribute only weakly to the final photoemitted intensities. At the same time, they also allow the R-A order to be adjusted at each stage in a scattering path, a unique feature not utilized before in PD simulations. Both options are controlled by one criterion, called pathcut, which is a cutoff criterion with value << 1. In this section, we indicate how this cutoff has been implemented and explore the resulting compromises between time savings and accuracy.
The pathcut criterion is applied as follows. Before
starting a multiple-scattering calculation, all individual
single-center scattering events involving a three-atom
vertex a ®
b ®
c and represented by a given
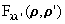 of the type shown in Fig. 2 are evaluated
separately. The largest value of
of the type shown in Fig. 2 are evaluated
separately. The largest value of
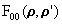 is taken as the reference value. All those
elements that are smaller than a factor
pathcut times this largest are
declared to be negligible; this is done dynamically in a
multiple-scattering path, so that scattering events further down
a path, which are normally weaker because of decay with distance,
will be cut off relatively more than early scattering events.
In this way, a scattering matrix is automatically reduced as
appropriate to a lower-order R-A event with smaller matrix size
and faster computation. In particular, if all elements of a
matrix are declared negligible, the path is terminated. Because
single scattering usually dominates, single scattering paths are
calculated using pathcut = 0.
is taken as the reference value. All those
elements that are smaller than a factor
pathcut times this largest are
declared to be negligible; this is done dynamically in a
multiple-scattering path, so that scattering events further down
a path, which are normally weaker because of decay with distance,
will be cut off relatively more than early scattering events.
In this way, a scattering matrix is automatically reduced as
appropriate to a lower-order R-A event with smaller matrix size
and faster computation. In particular, if all elements of a
matrix are declared negligible, the path is terminated. Because
single scattering usually dominates, single scattering paths are
calculated using pathcut = 0.
To illustrate the effects of pathcut for a typical large-cluster case, we show in Table 4 a summary of results for multi-layer emission from an 86-atom Cu(111) cluster at an energy of 100 eV, including the statistical weights in percent of scattering-amplitude matrix sizes when pathcut = 0.001 (a typical value that we have found to represent a good compromise between computation time and accuracy). Although second-order multiple scattering requires dealing in about 16% of the cases with 3rd and 4th order R-A or matrices of (10x10) and (15x15) size, for 3rd and higher multiple scattering order, 2nd order R-A and (6x6) matrices are found to be fully adequate. In fact, for 5th or higher multiple scattering order, 1st order R-A is probably adequate. To further quantify the effect of pathcut on the quality of the resulting photoemitted intensities, we define an intensity reliability factor RI as
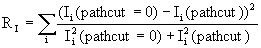
|
(39) |
where Ii represents photoemission intensities, and the sum runs over all the available data points for different energies or angles. Thus RI = 0 represents a perfect calculation, as defined by pathcut = 0.
| MS order | R-A order (matrix size) | |||||
|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | ||
| none | (1x1) | (3x3) | (6x6) | (10x10) | (15x15) | |
| 1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 100.0 |
| 2 | 0.0 | 1.1 | 39.3 | 43.2 | 7.6 | 8.7 |
| 3 | 33.6 | 53.5 | 12.1 | 0.7 | 0.1 | 0.0 |
| 4 | 27.3 | 32.3 | 5.3 | 0.5 | 0.0 | 0.0 |
| 5 | 61.9 | 20.1 | 3.0 | 0.3 | 0.0 | 0.0 |
| 6 | 83.8 | 13.7 | 2.2 | 0.3 | 0.0 | 0.0 |
| 7 | 87.6 | 10.3 | 1.8 | 0.3 | 0.0 | 0.0 |
| 8 | 89.7 | 8.3 | 1.7 | 0.3 | 0.0 | 0.0 |
| Table 4. Distribution (in percent) of different scattering amplitude matrix sizes as a function of multiple-scattering order (MS order) for an 86 atom Cu cluster with pathcut = 0.001, using default values at energy E = 100 eV. The column labeled none represents weak events that terminate a path. |
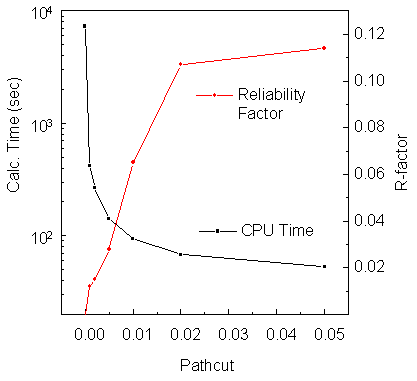
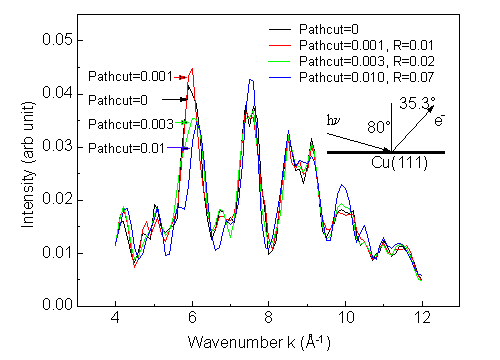
Fig. 11 shows the intensity reliability and calculation time as a function of pathcut for an energy scan of Cu 3p intensity over the energy range 60 eV to 550 eV for our 86-atom Cu(111) cluster. It can be seen that setting a pathcut value of 0.001 can easily save an order of magnitude of computation time compared to the full calculation without cut (pathcut = 0), and also that a great deal of time gain is achieved even with this small a cutoff criterion. That is, going from 0 to 0.001 gains by a factor of about 19, whereas going from 0 to 0.01 gains by about 75. Fig. 12 gives us a feeling for the quality of curve-to-curve comparisons at different pathcut values.
|
| Fig. 11. Intensity reliability factor RI (Eq. (39)) and calculation time on a 200MHz Sun Sparc Ultra-2 workstation as a function of pathcut, for various choices of this parameter in second-order R-A scanned-energy calculations for the 86-atom Cu(111) cluster. A value of 0 for pathcut corresponds to inclusion of all scattering events. |
|
| Fig. 12. Curve-to-curve comparisons of scanned-energy calculations for the 86-atom cluster and for different values of pathcut. Default values are used for other parameters. |
From this analysis and other calculations, we find that a pathcut value of 0.001 is fully adequate for the quantitative modeling of photoelectron diffraction data.
If a cluster is to represent an infinitely extended surface and/or include multilayer emission from a bulk specimen (as in the Cu case considered here), its size must be chosen large enough. To properly scale this problem, photoelectron waves leaving an emitter in free space decay in intensity with the inverse square of the distance from the emitter, i.e. as 1/r2: if there were no other damping effects, this would require an infinitely large cluster, since the number of scatterers on a shell at a given distance increases with the square of that distance, compensating the 1/r2 decay. Inelastic scattering adds an exponential decay factor, described theoretically by the inelastic attenuation length, which ensures that a finite cluster suffices. Vibrational effects and angular broadening act to further shrink the volume that is effective in producing diffraction modulations.
Fig. 13 again shows scanned-energy results for the ideal clean Cu(111), but this time calculated for clusters of different sizes. The photon polarization angle is again 10° off-normal and the intensities are taken from the Cu 3p core level in the direction of normal emission, allowing emission from all layers in the cluster. No pathcut is considered. To better compare these scanned-energy curves, we plot the usual c(k) curves defined as
| c(k) = (I(k) - I0(k)) / I0(k) | (40) |
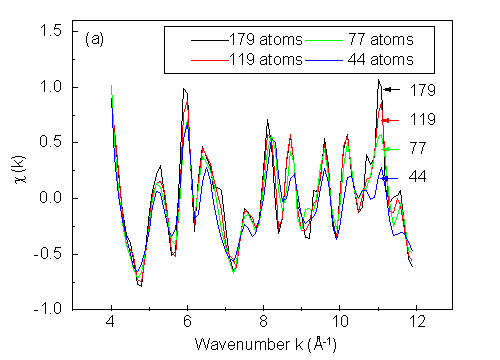
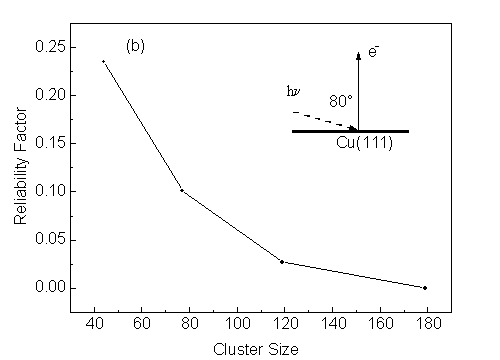
where I(k) is the photoemission intensity at wave number k, and I0(k) is the background subtracted from the intensity vs. wave number curve by using a spline fitting method. From Fig. 13 we can see that a 119-atom cluster yields most peaks and valleys at proper locations. Larger clusters become necessary for finer details. However, in practice, other effects not included here favor the sufficiency of smaller clusters: namely, vibrational damping of diffraction, and the experimental angular aperture (typically ±3° to ±5°), both of which will tend to smooth out fine structure.
Thus, we conclude that clusters of about 100 atoms in size should be sufficient for most problems, in agreement with prior studies.
|
| Fig. 13. (a) Effect of cluster size on scanned-energy calculated photoelectron diffraction c(k) curves for Cu(111), with clusters sizes of 44, 77, 119, and 179 atoms. Default values are used for other parameters. (b) Reliability factor as a function of cluster size, with the 179-atom result used as the reference. See text for further details. |
Finally, we consider the reliability with which such R-A calculations can be used to determine atomic structures, using the classic approach of theory-experiment comparison via reliability factors or R-factors. Although various definitions of R-factors exist, we will here use a rather straightforward definition of the goodness of fit between theory and experiment for photoelectron diffraction data:
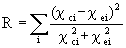
|
(41) |
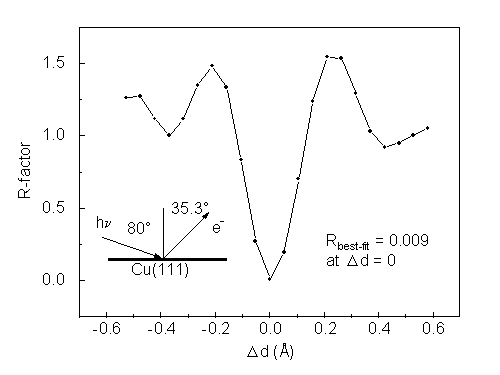
where cci and cei are calculated and experimental c curves, respectively (c.f. Eq. (39)). As one way of estimating the reliability of the Rehr-Albers approximation for structure determinations, we have replaced the experimental data with the calculated scanned-energy (60 to 550 eV) results from the exact formalism, which is based on a smaller 35-atom cluster representing Cu 3p emission from ideal clean Cu(111). We have used our R-A method with R-A order only up to 2 (6x6 matrices) to explore sensitivity to possible variations in the outermost interlayer spacing by calculating the same scanned-energy curve and quantifying the fit to the exact result. The variation of R with interlayer spacing is shown in Fig. 14. It indicates a best fit R value of 0.009 at precisely the interlayer spacing that was used in the exact calculation, thus giving confidence in the ability of this method, even at 2nd R-A order, to accurately determine structure, while saving computer time. Our current implementation of the R-A method with pathcut and adjustable orders should do even better than this.
|
| Fig. 14. Assessment of atomic position reliability of photoelectron diffraction calculations using the second-order R-A approximation, shown by varying the first interlayer spacing and comparing to intensities calculated by using the exact formalism with zero interlayer relaxation. A cluster of 35 atoms was used. |
The Rehr-Albers (R-A) separable propagator approximation up to 4th order (and using up to (15x15) matrices) has been applied to the calculation of photoelectron diffraction curves. By replacing the propagator matrices in the exact Green's function formalism by the much smaller scattering-amplitude matrices of R-A, this approximation saves much computation time. Our convergence tests for typical conditions in photoelectron diffraction indicate that 4th order R-A is highly accurate for all cases likely to be encountered. Furthermore, 2nd-order R-A (with (6x6) matrices) applied with clusters of 100 or more atoms and at least 7th-order multiple scattering, and using a pathcut of about 0.001, provides excellent results within 5% of exact results for most cases, particularly if the initial state is of s or p type. Higher Rehr-Albers orders are necessary for the first scattering events involved with initial states of d type (3rd order) and f type (4th order), but can be neglected in later events. We have also implemented R-A in a program which automatically adjusts the R-A order from 4th downward according to the pathcut criterion, and this should permit fully quantitative and maximally efficient calculations for any situation. At least an order of magnitude in computation time is saved by recognizing that lower orders of Rehr-Albers suffice for most higher-order multiple scattering events. Larger clusters may be necessary for describing all fine structure in diffraction curves, but approximately 100 atoms should be the maximum needed for most cases. Looking ahead to future applications of the Rehr-Albers method, we note that several simulations of actual experimental data, e.g. on surfaces of W(110), O/W(110), Li/Al(111), and MnO(100), etc., have also been performed using MSCD.
| Return to Van Hove home page |
|---|