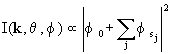
The technique of core-level photoelectron diffraction (PD) has been applied to surface structure determination on the atomic scale for more than 20 years. This local diffraction technique probes short-range order around the photoemitter. A broad variety of surfaces have been successfully studied, including metals, semiconductors, oxides, systems exhibiting surface core-level shifts, adsorbed atoms and molecules, epitaxial overlayers, and atoms at buried interfaces. A number of research groups have performed photoelectron diffraction experiments to study surface and interface structures, using both laboratory X-ray and synchrotron radiation sources, and doing the measurements in both scanned-angle and scanned-energy modes. Several reviews of this field have appeared in recent years. A further element which has recently been added to this technique is the application of holographic inversion methods to photoelectron diffraction data so as to more directly yield atomic structures in three dimensions. Thus, the accurate theoretical modeling of photoelectron diffraction is crucial to the development and use of this technique in its various forms, and we here discuss one method for accomplishing this.
In core-level photoemission, a photon illuminates an emitting atom at or near the surface or in the solid and excites an electron from an atomic core-level, ejecting the electron to a detector far away from the surface. The resulting photoelectron wave components can occur both by direct propagation to the far-field detector and via a number of scatterings from atoms in the neighborhood of the emitter. The quantum interference (diffraction) between the different pathways depends sensitively on the relative atomic positions. The intensity associated with this process, when measured as a function of photon energy and/or emission angles, thus includes information about the atomic structure around the emitter. The energy- and angle-resolved photoemission intensity I(k,q, f) at the detector can be written in general as
|
|
(1) |
where k is the final electron wave vector (related to the final kinetic energy), q and f are the polar and azimuthal angles of photoelectron emission, respectively, f0 is the wave-component representing travel along a path directly to the detector without being scattered by another atom, and fsj is the wave-component representing travel via paths involving single or multiple scattering by one or more atoms, respectively. The multiple-scattering order is defined as the number of scattering atoms in a path: j = 1 = single, 2 = double, etc. Higher-order scattering processes are less important because of the roughly 1/r falloff of the outgoing distorted spherical wave represented by f0, damping due to inelastic processes and loss of diffraction modulation due to thermal vibrations (Debye-Waller effects). The explicit sum in Eq. (1) includes all possible scattering paths inside the solid.
To briefly review the history of such theoretical modeling of PD effects, the first qualitative analyses of higher-energy data made use of Kikuchi-band theory. An early quantitative theoretical explanation of these diffraction data was provided by Liebsch in 1974 and then improved by him and others with multiple-scattering effects in 1976; it was based on LEED theory. In the next few years, Pendry, as well as Li, Lubinsky and Tong, put forward similar theories involving the more complex nature of both the initial and final states. These theories require the assumption of full translational symmetry parallel to the surface for the system under investigation.
A single-scattering cluster-based theory based on prior work in extended x-ray absorption fine structure (EXAFS) and preliminary work on Auger electron diffraction (AED) was first applied to scanned-angle photoelectron diffraction by Kono et al; this was based on the plane-wave approximation. The next major advance in the cluster approach was by Barton and Shirley, who included both spherical-wave corrections and multiple scattering effects. The cluster-based theories are inherently more suitable for photoelectron diffraction modeling in view of the point-source nature of the problem, the spherical outgoing waves involved, the short inelastic attenuation lengths, and the sensitivity to short-range rather than long-range order. In particular, one key advantage of PD as a structure probe is in not requiring long-range translational order, and so being able to model such short-range order structures conveniently without having to resort to some sort of repeated structure with fictitious long-range order is crucial. Cluster-based methods are ideal for this. It has also been found that the maximum cluster size required for accurate simulations is about 100 atoms.
The multiple scattering expansion for spherically-symmetric
scatterers is conveniently formulated in terms of diagonal
plane-wave scattering t-matrices with elements
tl = sin(dl)exp(i
dl)
and matrix elements of the free-particle propagator
GL,L'(r) =
<L,R|G|L',R'>
in an angular momentum L = (l,m) and site R basis.
Here r = k(R-R')
denotes the dimensionless vector between two sites, k = |k|
is the wave number.
GLL'(r)
is defined by the following integral involving spherical Bessel
functions jl(kr) and spherical harmonics
YL(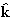 ):
):
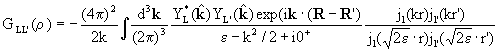
|
(2) |
where is a unit vector along k,
r and r' are arbitrary displacements. An (n-1)-atom path (Fig. 1),
including the emitter atom at R0, n-1 scatterers at
R1, R2, ...,
Rn-1, and the detector at Rn,
is represented by the following total propagator in this
exact multiple-scattering expansion:
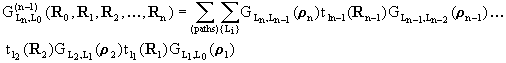
|
(3) |
where ri = k(Ri+1-Ri), and L0 and Ln denote fixed initial and final angular momenta.
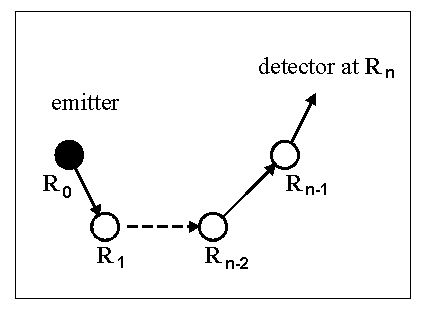
|
| Fig. 1 A photoelectron excited from the core-level of an emitter atom at R0 propagates to the detector at Rn via n-1 scatterers R1, R2, ..., and Rn-1. Each straight segment linking consecutive atoms represents a Green's function propagator GLL'(k(Ri+1-Ri)). The emitter can become a scatterer after emission. |
Several computer programs have been written to successfully calculate photoelectron diffraction intensities based on this exact cluster formalism, for example, by Chassé and co-workers, and by García de Abajo. However, without special optimization (as we consider below) such exact programs in general require considerable computing times for large clusters, due to the large matrix dimensions involved and the large number of paths that are explicitly summed over. That is, the matrix GLL' has dimensions (lmax+1)2 x (lmax+1)2, where lmax is the largest angular momentum value needed to accurately represent scattering; it can be roughly evaluated using lmax ~ kmaxRmt, where Rmt is the muffin-tin radius of the scattering potential. For photoelectron diffraction in the energy range 50-1500 eV, this lmax lies between 5 and 30, respectively, of course depending on the electron energy. The computing time can be considerably reduced by an iterative summing over the multiple-scattering paths, as proposed by García de Abajo, which leads to proportionality to the number of multiple scatterings, rather than the much steeper behavior as in the traditional summing method. More precisely, the total calculation time in traditional summing is T µ T2 nmax/2, while the iterative summing has a total time of T µ nmaxT2, where T2 is the time for a double scattering calculation, and nmax is the maximum multiple-scattering order included. A convergent calculation is found to requires nmax up to about 6, or even higher in cases with long straight chains for strongly forward scattering atoms. We use these two exact formalisms here as references with which to compare calculations performed with the Rehr-Albers expansion.
Several methods have been proposed previously for approximating the exact scattering formalism so as to reduce computation times in cluster calculations.
By applying object oriented programming within C++, the MSCD code provides a better user interface (particularly important for convenient use by experimentalists), general data formats, and fully functional subsidiary utilities. The simulation program is also designed to be very portable, with very simple changes from one computer type to another. The sequential version of the code has been implemented on supercomputers (under Unix), on Sun workstations (Unix), as well as on PC and Macintosh desktop computers. The parallel version has been implemented on a massively-parallel supercomputer (Cray T3E) and on a cluster of multiprocessor systems (COMPS, a type of network of workstations) under MPI.
Comprehensive test calculations agree well with more exact (but much slower) calculations performed by other programs. A new version is currently being generalized to take into account photon and electron-spin polarization effects such as circular dichroism (the latter is obtained by contrasting measurements performed with right- vs. left-circular polarized light), magnetism (including magnetic circular dichroism), and relativistic effects such as spin flip.
In the next sections, we will briefly introduce both the exact and R-A formalisms, discuss the methods that have been used to increase the calculational efficiency of R-A, present a number of results calculated with the MSCD program based on R-A, and compare these results with exact cluster calculations to quantitatively evaluate the performance and limitations of the R-A approximation as carried out to different orders. We will discuss a few more necessary details about the exact cluster-based formalism, and briefly review the R-A method and its properties. The methodology and convergence of the implementation of R-A used in the MCSD will be presented, as well as the overall reliability of the MSCD approach for structural determinations.
| Code | mscd | scat | txpd | newchp | spdl | javier |
| Version | 1.37/98 | 3.65/96 | 95 | 91 | 97 | 97 |
| Language | C++ | Fortran | Fortran | Fortran | Fortran | C |
| Method | R-A approx | R-A approx | R-A approx | JB approx | Exact | Exact |
| Platform | PC/Mac/Sun/ Cray/Comps |
PC/Mac/Sun/ Cray |
Sun/Cray | Mac | Sun/Cray | Sun |
| Parallel | Yes | No | No | No | No | No |
| Ini. State | all | all | all | S only | all | all |
| Scanning | energy/angular | energy/angular | angular | energy | energy/angular | energy |
| Rotation | analyzer/sample | analyzer/sample | sample | N/A | analyzer | N/A |
| Hologram | Yes | Yes | Yes | No | No | No |
| Fitting | Yes | Yes | No | No | No | No |
| Vibration | correlated | correlated | correlated | uncorrel. | uncorrel. | uncorrel. |
| Polarization | linear | linear | linear/circular | linear | linear/circular | linear |
| MLD/MCD | No | No | Yes | No | Yes | No |
The programming of the MSCD package began on December 9, 1996, using the ANSI C++ object oriented programming (OOP) technique and message passing interfacing (MPI) parallel communication.
We thank Dr. H. Wu, Dr. Z. Hussain and D.A. Shirley for their collaboration in developing the SCAT package. The MSCD package uses the same method and similar data format, and this documentation takes some contents from the SCAT package user guide.
We thank our colleagues in D.A. Shirley's group at Lawrence Berkeley National Laboratory, E. Moler, S. Kellar, W.A. Huff, X. Zhou, and B. Peterson, for their detailed discussions and many test calculations. Special thanks also go to C.S. Fadley, J.J. Rehr and A. Kaduwela for many helpful discussions regarding photoelectron diffraction theory and its computation. R.X. Ynzunza, F.J. García de Abajo and A. Chassé ran many time consuming test calculations and comparisons with other codes, which helped us to debug and improve the software.
| Return to Van Hove home page |
|---|