We have developed a program package to calculate and analyze the photoelectron diffraction intensity and their chi function using C++ with Object Oriented Programming (OOP) and the parallel computing technique Message Passing Interface (MPI). The code features the multiple scattering approach developed by Rehr and Albers, the TPP-2 inelastic mean free path formula developed by Tanuma, Powell and Penn, and the correlated temperature effect developed by Sagurton, Bullock and Fadley. The code simulates various kinds of experimental scanning modes, sample or analyzer rotation mechanisms, chi calculation algorithms, and their combinations. The scanned-angle calculation and built-in fitting procedure fits most analysis needs of photoelectron diffraction data. By avoiding any repeat calculations for identical events and identical path summing, the program speeds up the calculation dramatically compared with several other codes.
To run the program on different platforms, we split our package into two parts: a machine independent source code; and a machine-dependent source code. The same machine independent source code applies to both sequential and parallel computers. Using this structure, we have maximized code portability. Because the machine-dependent source code is relatively quite small (about 2 Kb), we can easily rewrite it to transfer the package to a new kind of computer.
The following diagram sketches the division of the code into a machine-independent part ("core program") and a machine-dependent part ("shell functions"). It gives examples of general calls in the core program that use machine-specific calls in the shell-functions part.
| Parallel platforms | Sequential platforms | |
|---|---|---|
| Cray T3E, COMPS | Cray J90, Sun workstation, IBM PC and Macintosh | |
|
Core Program (Machine Independent) |
Shell Functions (Machine Dependent) |
|
| 450 K source code, same for parallel or non-parallel | 2 K source code for each platform | |
| Use machine independent function calls everywhere |
|
CPU time usage, and basic MPI functions |
Example:
int main(int argc, char **argv)
{ mpiinit(argc,argv);
begcputime=processortime();
... ... ...
endcputime=processortime();
mpiend();
return(error);
}
|
|
Example for Cray T3E:
long processortime()
{ return(ta=times(&timebuf)); }
void mpiinit(int *argc,char ***argv)
{ MPI_Init(argc,argv); }
void mpiend()
{ MPI_Finalize(); }
|
|
Library functions, taken care of by C++ compiler Example: Void MPI_Init(argc,argv); |
||
| To perform scanned-energy calculation or fitting | |
|---|---|
| executable file | mscd |
| batch input file | mscdin.txt |
| include files | input files, phase shift files, radial matrix files, experimental scanned-energy data files (for fitting only) |
| output files | calculated scanned-energy data files |
| To calculate phase shift and radial matrix data files | |
| executable file | psrm |
| input file | psrmin.txt |
| include file | potential data file |
| output file | phase shift or radial matrix data files |
| To perform real space hologram transformation | |
| executable file | holo |
| input file | holoin.txt |
| include file | calculated or experimental scanned-energy data file |
| output file | real space image data |
| To calculate chi from experimental or calculated intensity data | |
| executable file | calchi |
| input file | scanned-energy data with intensity |
| output file | scanned-energy data with intensity and chi |
| To calculate normalized chi from experimental or calculated data file | |
| executable file | calnox |
| input file | scanned-energy data file |
| output file | scanned-energy data file |
| To calculate difference between two scanned-energy data | |
| executable file | caldif |
| input file | two scanned-energy data file |
| output on screen | reliability factors |
| To convert potential data to mscd format | |
| executable file | poconv |
| input file | simple format potential data (two columns) |
| output file | mscd format potential data |
| To convert phase shift data to mscd format | |
| executable file | psconv |
| input file | Fadley's group format phase shift data |
| output file | mscd format phase shift data |
| To convert radial matrix data to mscd format | |
| executable file | rmconv |
| input file | Fadley's group format radial matrix element data |
| output file | mscd format radial matrix element data |
| To calculate IMFP using TPP-2 formula or attenuation length | |
| executable file | calmfp |
| input on screen | follow instructions on screen |
| output file | mscd format mean free path data file |
| To calculate thermal vibrational mean square relative displacement | |
| executable file | calvib |
| input on screen | follow instructions on screen |
| output file | mscd format vibrational MSRD data file |
| To calculate photoelectron scattering factor | |
| executable file | calfac |
| input on screen | follow instructions on screen |
| output file | mscd scattering factor data file |
All the data files used or output by the mscd package programs are ASCII text file, having a unified mscd format data structure. This format consists of three parts.
Commonly used symbols and their definition in the mscd package:
| symbol | description |
|---|---|
| k | photoemission wave vector in unit of Å-1, k=0.512331*sqrt(kinetic energy in eV) |
| theta | photoemission detector polar angle in unit of degree |
| phi | photoemission detector azimuthal angle in unit of degree |
| I | angle-revolved photoemission intensity, in arbitrary unit |
| I0 | intensity background, in arbitrary unit |
| I0t | intensity reference, i.e. the calculated photoemission intensity contributed by the emitters only without scatterers |
| I0b | intensity background, i.e. a smooth function extracted from the calculated or experimental intensity data by using weighted sample spline fitting (for k or theta dependent curve), or a constant which is the average of the intensity data over all the azimuthal angles (for phi dependent curve only) |
| chi | non-dimensional function chi=(I-I0)/I0 |
| l | angular momentum index |
| lmax | maximum value of angular momentum index |
| m | magnetic quantum number |
| li | initial angular momentum |
| lf | final angular momentum ( lf=li-1 and li+1 ) |
| delta(l) | phase shift of a scattering event for angular momentum l |
| R(lf) | amplitude of the radial component of the dipole matrix element into the given final state (lf) |
| delta(lf) | phase of the dipole matrix element into the given final state (lf) |
Definition of the three-digit datatype of scanned-energy data file:
| name | definition | |
|---|---|---|
| third digit from left | 1 | energy dependent curves or hologram in (k I I0 chi) |
| 2 | polar angle dependent curves in (theta I I0 chi) | |
| 3 | azimuthal angle dependent curves in (phi I I0 chi) | |
| 4 | solid angle dependent hologram in (theta phi I I0 chi) | |
| 5-8 | same as 1-4, but normalize chi | |
| second digit from left | 1 | rotations of the geometry are made by rotating the analyzer in both polar and azimuthal directions |
| 2 | rotations of the geometry are made by rotating the sample in both polar and azimuthal directions | |
| 3-6 | reserved for MCD implementation | |
| first digit from left | 1 | calculated photoelectron diffraction pattern, chi is calculated by theoretical result chi=(I-I0t)/I0t |
| 2 | calculated photoelectron diffraction pattern, chi is calculated by theoretical result chi=(I-I0b)/I0b | |
| 3 | experimental photoelectron diffraction pattern, chi is calculated by experimental data chi=(I-I0b)/I0b | |
| 4 | same as 3, but no intensity I and background I0 data | |
| 5-6 | reserved for MCD implementation |
Date types of other files:
| 711 | phase shift data in (k delta(l=0 1 2 ...)) |
| 721 | radial matrix data in (k R(li+1) delta(li+1) R(li-1) delta(li-1)) |
| 731 | calculation report |
| 741 | input data file for photoemission calculation |
| 751 | a batch of input files (filename should be mscdin.txt) |
| 811 | potential data in (r (angstrom) rV (eV-angstrom)) |
| 821 | input file for phase shift or radial matrix (psrmin.txt) |
| 831 | subshell eigen wave function data in (r (angstrom) u (arb unit)) |
| 911 | real space hologram image file in (x y z (angs) intensity (arb unit)) |
| 921 | input file for hologram transformation (holoin.txt) |
| 931 | effective scattering factors as function of energy and scattering angle |
| 941 | electron inelastic mean free path data as function of energy |
| 951 | thermal vibrational mean square relative displacement data as function of bond length |
Unit cell and cluster selection
The most complicated text file in mscd package is the input data file for a scanned-energy calculation. This file consists of non-structural parameters (like scanning mode, fitting mode, initial state, geometry, inelastic mean free path formula, temperatures, etc.) and structure parameters which determine the locations of atoms.
We define the surface structure by logical layers. A logical layer is a two-dimensional primitive lattice, or a two-dimensional Bravais lattice, which specifies the 2D-periodic array. A non-primitive physical layer must be divided into 2 or more primitive logical layers. Thus, a logical layer consists of all atoms with position vectors R of the form R(x,y,z) = m a(x,y) + n b(x,y) + Rlayer-origin(x,y,z), where a and b are two-dimensional vectors, m and n range through all integral values which can be negative, zero as well as positive. Rlayer-origin(x,y,z) is the location of the sample origin. The choice of two-dimensional vectors is not unique.
If a physical layer consists of two kinds of atoms, the layer can be divided into two logical layers. Each logical layer can have only one kind of atom. If the atoms in a physical layer are emitters, basically all the atoms in that layer are emitters, by periodic equivalence. Since the photoelectron diffraction intensities from different emitters are independent, we only need to consider a few emitters with different neighboring scatterers. In our code of photoelectron diffraction calculation, we take only one emitter for each logical layer: it is the atom which sits at that layer's origin. This way, even a primitive emitter layer may have to be divided into several logical layers, if more than one emitters have to be considered because of different neighboring atoms in three dimensions. This is the case, for instance, when an overlayer has a 2D structure with a superlattice or disorder, making substrate atoms translationally inequivalent: then the emission from these different atoms must be calculated independently and summed.
The ideal structure of a surface is infinite in extent in two dimensions and infinitely deep. To correctly represent photoelectron diffraction, we must in principle therefore take account of all scatterers within the surface. In practice, the contribution from a scatterer far from the emitter may be ignored. So we only need to choose a finite cluster including a few emitters and a finite number of scatterers, typically 100 atoms. In this program package, we use a cluster shape defined by a semi-ellipsoid: the profile as seen parallel to the surface is a semi-ellipse of minor axis r and major axis h; perpendicular to the surface it has as cross-section a circle of radius r. All the atoms within this semi-ellipsoid will be taken into account in the calculation. All the atoms outside this cluster are ignored. A bigger cluster selection will give a more accurate calculation, because of the smaller edge effect. But a bigger cluster calculation will take much more computation time and require a much larger computer memory. We have to make a compromise between cluster size and computation time. Typically, a cluster of 80 to 100 atoms is a reasonable choice, which requires 10 MB to 16 MB of random memory for single precision floating point computation, or double that for double precision floating point processing.
For testing purposes, we have two other alternative choices for the cluster. We can draw different circles or rectangles for each logical layer, all the atoms on that layer within that circle or rectangle will be considered.
Non-structural parameters
The non-structural input parameters are initial core-level,
number of multiple scattering orders, R-A approximation
order, scanning mode (description of energy scanning,
polar angle scanning, azimuthal angle scanning, or hologram,
i.e. two-angle scanning,
using the same codes for the data type), detector instrumental
half-aperture angle, photon polarization angles, Debye
temperature, density and atomic weight for each kind of
atoms, and number of valence electrons for the inelastic mean
free path calculation.
See detail of those definitions.
In the current version, we only allow linearly polarized photons (other light polarizations are linear combinations of two perpendicular linear polarizations, and can be produced as such). The direction of photon polarization is described by its polar and azimuthal angles, theta(p) and phi(p). They are defined as the polar and azimuthal angles with respect to a reference direction, which depends on the rotation mechanism of the experimental geometry. If the surface normal direction is fixed (for example do not rotate sample, or only rotate it in azimuthal direction), the reference direction is this fixed surface normal. If the surface normal is not fixed (rotatable), in a reasonable experimental geometry, the analyzer must be fixed, then the reference direction is this fixed analyzer direction.
The photoemission intensity curves versus energy or angles consist typically of a series of diffraction peaks and troughs. To compare calculated with experimental intensities I, we have to remove the atomic partial cross section from intensities I0, leaving only the oscillating part of the intensity, which we call chi function or c function.
chi = (I-I0)/I0
Because the free atom cross-section I0 contains only very low frequency information, we will make little error at the structurally important frequencies if we approximate I0 as the smooth part of I.
More specifically, in our MSCD program package, to extract this background I0 from an energy-scanned or polar-angle-scanned curve, we fit it as a cubic spline function to both calculated and experimental intensity curves using the non-linear Marquardt fitting method. To make this spline function, we choose 3-5 initial data points of equal step as fitting parameters, which ensure a smooth background and have only very low frequencies. For an azimuthal-angle-scanned curve, because the cross-section along azimuthal direction can be thought as a constant, we just use the average of the intensities as the background I0.
Although the multiple scattering theory can well reproduce the experimental photoemission intensity, the agreement is never perfect. To quantify the goodness of fit or to automatically fit structural parameters to the data, we introduce the reliability or R-factor. In our MSCD package, we use two reliability factors Ra and Rb, defined as follows together with a similar factor R:
R = sumj(chicj - chiej)2 / sumj(chiej)2
Ra = sumj(chicj - chiej)2 / sumj(chicj + chiej)2
Rb = sumj(chicj2 - chiej2) / sumj(chicj2 + chiej2)
These R-factors are related to the traditional Pendry reliability factor for used in LEED; chic is the calculated chi value, while chie is the experimental one, the sum index j is taken over all the corresponding data points for both curves. The first factor R is used by many groups, Ra is the factor used in our fitting procedure, Rb is a factor which tells us the quality of amplitude agreement. The largest effect on the overall amplitude of the chi function stems from the thermal vibration: thus, from the Rb value, we will have an idea how good the Debye temperature is.
In the complicated photoelectron diffraction theory, many parameters, such as temperature, inner potential, geometry and structural parameters, affect the photoemission intensity in a non-monotonous way. Thus, fitting may be the only effective method to perform the fitting of such parameters to experiment. A good reliability factor is then required to control the fitting process. In our MSCD package, we use the Ra factor, instead of R, to peform the fit, for the following reason.
For the best trial calculation, R and Ra are both small (<< 1.0). The amplitudes of calculated and experimental data are quite close, i.e.
Ra-best ~ Rbest
For the worst trial calculation, the calculated intensity is irrelevant to the experimental curve. We would have
Rworst =
1 + Atry2/Aexp2
Ra-worst = 2.0
Here, Atry and Aexp are the average amplitude of the trial calculated and experimental intensity curves, respectively. Because Atry could be much less than Aexp in the fitting procedure while changing parameters, especially the Debye temperature, so we can write
Rworst = 1.0
Ra-worst = 2.0
Using the simplest single-scattering model of scanned-energy, chi can be written as
chi(k) = sumj { Aj(k) cos[ k (Rj - Rj cos(thetaj) + phij ] }
where k is the wave vector, Aj the amplitude, Rj the bond length, thetaj the scattering angle, and phij the phase of the jth scattering event. Let us consider only one component with same frequency k but with different amplitudes and phases for the trial and experimental curves, namely
chic(k) = Atry cos( k x + phi)
chie(k) = Aexp cos ( k x )
where x = R(1-cos(theta)), and chic and chie are calculated and experimental chi curves. From the R-factor definitions, we obtain
R / Rworst = [ Atry2 +
Aexp2 - 2 Atry
Aexp cos (phi) ] /
Aexp2
Ra / Rworst = [ Atry2 +
Aexp2 - 2 Atry
Aexp cos (phi) ] /
[Atry2 + Aexp2]
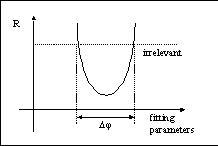
Here we found Ra/Ra-worst < R/Rworst. We know, once a trial R-factor is greater than one of the extreme worst cases, that the fitting procedure would fail to continue towards the best minimum. The next figure shows an R-factor plot versus the fitting parameters. Assuming Atry = Aexp, delta(phi) in the figure will be the available fitting range of phase phi in the above equation. Obviously, we hope phi to be as large as possible. From the above equation, we find phi = 120° if we use R as R-factor and assume Atry = Aexp, and phi is even smaller when Atry < Aexp. However, we can see that phi = 180° if we use Ra as the R-factor to monitor the fitting process. This is the reason we use Ra as the monitoring R-factor in our fitting procedure.
|
Figure. R-factor plot versus fitting parameters. If a trial R-factor is greater than the worst case (irrelevant), the fitting procedure would fail to continue. |
|---|
For a poorly-known system, we may have several structural parameters to fit, such as layer spacing, location of adsorbate atom, surface reconstruction, etc. Because the inner potential and Debye temperature are not well understood so far, we have to keep them adjustable, too. All these parameters influence the photoemission intensity in a complicated way. Changing any of them may introduce more than one local minimum. Therefore, a simple conventional fitting method would not be enough in most cases. In our code, we perform the fitting in a three-step process.
In the first step, we do a net search, in which calculations are made on a set of given grid points in fixed stepsize basis for each fitting parameter, and their Ra factor is monitored. This allows us to search a larger area, even if there are more than one local minimum. Ending this process, the best minimum and its parameters are chosen as the initial parameters of the next step.
In the second step, the program uses a downhill simplex method in multi-dimensions monitoring the Ra factor. This method requires only function evaluations, not derivatives. It is not very efficient in terms of the number of function evaluations that it requires. However, it may frequently be the best method to use if the figure of merit (here Ra factor) is "get something working quickly" for a problem whose computational burden is small. Once a trial calculation gives an Ra factor less than a given (input) tolerance, the downhill simplex loop will stop and transmit the best parameters to the next step.
| Return to Van Hove home page |
|---|